by: Samuel Chu, Jonathan Dong, Yiming Hao, Brianda Plascencia
The full report can be found here
Introduction
Health researchers are perpetually exploring novel methods to develop drugs for combatting evolving diseases. According to a statement in a 2016 article by Jim O’Neill, “By 2050, drug-resistant pathogens are expected to be the leading cause of death in the world.” A compelling instance supporting this statement is the emergence of COVID-19 and its variants, which have resulted in the deaths of millions worldwide. When considered alongside other significant diseases such as swine flu, Ebola, and HIV, it further underscores the urgency of developing drugs swiftly to mitigate their spread and adaptation.
Researchers have been exploring antimicrobial peptides (AMPs) as a potential avenue. AMPs are chains of amino acids that have displayed properties beneficial in combating pathogens. Identifying the presence of AMPs within these chains enables researchers to leverage them for the production of improved drugs or antibiotics. However, the structural complexity of AMPs makes the detection of new ones challenging and costly.
With the advent of deep learning models, researchers are turning to them for their ability to swiftly, efficiently, and accurately identify AMPs. In a paper titled “Identification of antimicrobial peptides from the human gut microbiome using deep learning,” by Ma et al., utilized a combination of deep learning models, such as long short-term memory model (LSTM), an attention model, and a bidirectional encoder representations from transformers model (BERT). Their aim was to assess the accuracy of these models in identifying AMPs based on an existing dataset comprising both AMPs and non-AMPs. Among all the combinations, an ensemble of the LSTM, attention and BERT models performed the best in identifying AMPs and non-AMPs. For our project, we aim in replicating and training solely on the attention model with new data to detect the presence of new AMPs accurately. Specifically, we would like to test the model’s performance on a dataset called FINRISK to determine its capability of detecting AMP presence within the collected data and establish any potential correlations with various conditions or diseases.
While previous studies have been done on AMP identification and on the FINRISK dataset, none of them examine the relationship between AMPs and health outcomes. The goal of this project is to provide a fast and efficient solution in identifying AMPs and use these results to possibly aid in the development of treatment or preventive measures for diseases in patients in hopes of expediting their recovery process.
Data
The dataset we utilized for this project is called FINRISK. It is a study conducted by the Finnish government which consists of DNA samples from the Finnish participants along with their health outcomes. This health data was recorded extensively on the study participants approximately 20 years later. We extracted 108 million DNA sequences from 71 participants within the database accessible exclusively through Barnacle2, the Knight Lab’s supercomputer cluster, due to the confidential nature of the human data within the FINRISK dataset. While we would have wanted to extract more patients’ data, we had to take this subset of patients due to time constraints.
Since our models input requires amino acid data, we would have to first convert the original data before we can use it. To achieve this, we developed a script capable of taking DNA inputs and generating their 6-frame translation. This process reads the DNA across three potential overlapping frames forward, as well as on the complementary strand in the reverse direction. The reason behind doing this is that it examines each nucleotide sequence in all six frames, forward and reverse readings, which allows us to identify all possible codons, including those that could start or stop protein synthesis. Thus, by translating these codons into their genetic code, we can decipher the sequence of amino acids encoded by the DNA. This makes possible the conversion of genetic information into functional proteins, which are composed of peptides–short strings of amino acids.
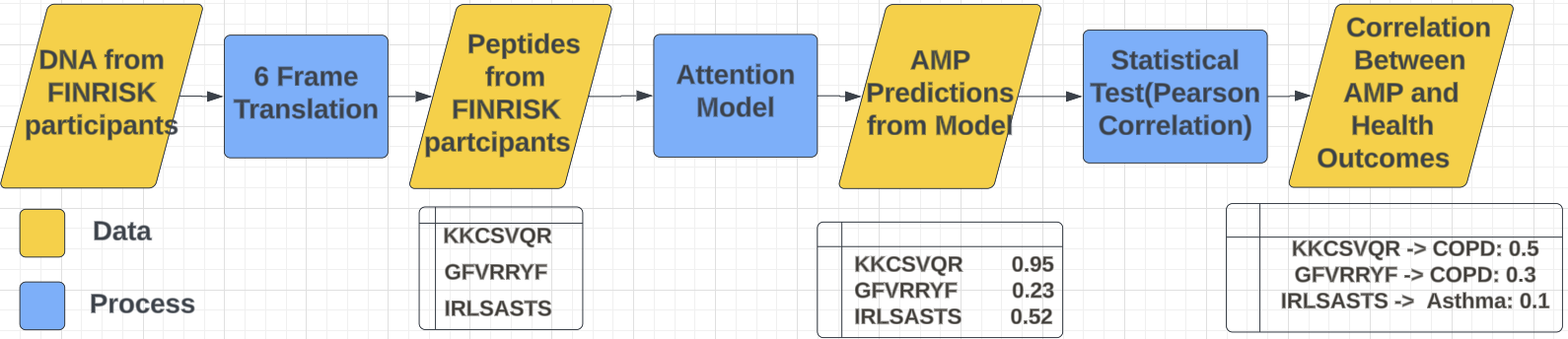
Figure 1. General pipeline of our experiment including data transformation
Methods
The model that we used for this experiment is the attention model, a neural network that is useful when it comes to processing text. As our input data consists of long strings of DNA sequences, the attention model would serve as the ideal neural network to perform our task on.
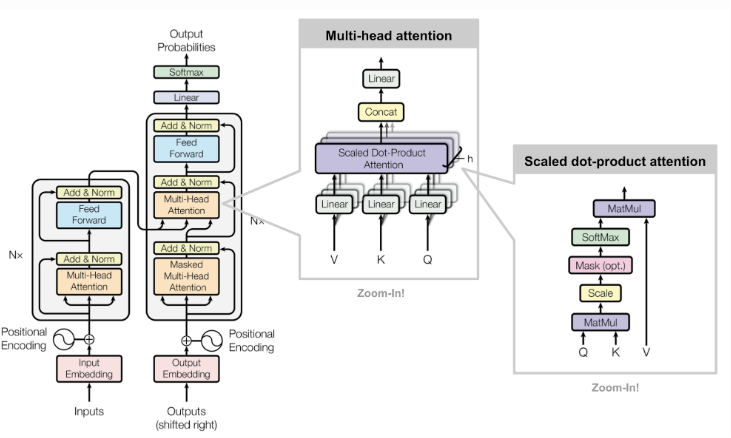
Figure 2. Framework of an attention model
After converting our original data into amino acid sequences, we performed tokenization to convert the data into an embedding of numbers corresponding to the amino acid. From there we would have to pad these sequences with 0’s to ensure each input is of equal length.
Our model would return the probability that the given amino acid sequence would be an AMP. To maintain accuracy in our predictions, we only kept the sequences that were 99.99% likely to be AMPs.
Once we have obtained a complete list of possible AMPs, we pulled the health conditons of the participants from the original dataset and looked to see if there was any correlation between them and our AMPs. In particular, we wanted to compare our AMPs to a few notable diseases or conditions such as Type 2 Diabetes (DIAB_T2), Chronic Obstructive Pulmonary Disease (COPD), Asthma, and High-Density Lipoprotein (HDL) disorders. Using a Pearson correlation test, a value between -1 and 1 that measures how strongly correlated two variables are, we were able to determine how certain AMPs are linked with these health problems.
Results
| Moderately Correlated (r>=0.5) | Lightly Correlated (0.3<r<0.5) | Negatively Correlated (r<0.0) | |
|---|---|---|---|
| DIAB_T2 | N/A | PYRKWCNNSCCVEGVAVWCPNCDNG AERIPRCDQQAAGQGCGRGVCRFRRCGGKAW WKWPGNSIRCSAVRRQTWYRSAWCCSAWTW |
N/A |
| COPD | GFVRRYFGYKSGILCRRGCVCRGWKRK IRLSASTSICKVSCTVSDKACCCSGGSLSNTGVCCSCKNSGTC CPAFSGHCHTPWGVCRPAMCRCAE KKCSVQRCTFSYAKKDGKCKGMFRVE |
N/A | N/A |
| ASTHMA | N/A | LWAVCRKVCRR CYRNRLCCSSCSKG IYGSFKRRFGCCHLRNTC |
N/A |
| HDL | N/A | CRIFKCIISICRK NYFRKGFCPRNECAVH YPIRGTCIKTFC |
RDLYRSNICCIRHGYC WRQGLCRWGGCR RPLQHRLQRFGKKIRRRNSCQVPS |
Representing our Pearson correlation coefficient as r, the above table displays the health conditions and some of the AMPs that are moderately correlated, lightly correlated and negatively correlated with them. Interestingly enough, the four AMPs that were moderately correlated with COPD were the only AMPs that displayed moderate correlation between all AMPs and the health conditions we tested. When we inquired about this with our mentor, they said that these peptides were novel discoveries and that this could prove to be an interesting thing to look into with more research.
There were many more AMPs that display some light correlation with these health problems that despite not being as promising as the 4 moderately correlated ones, can warrant further experimentation/research. In regards to HDL disorders, we also discovered some of the only instances where there were negatively correlated AMPs with the disorder.
It can be said however that the majority of AMPs that we have predicted show no correlation (r score of about 0) towards the health conditions. While these AMPs display no relation to our list of ailments, we used a small sample size of 71 study participants and 4 diseases. In the near future, we wish to conduct our analysis on more study participants and diseases.
Conclusion/Discussion
Through this project and its results, we wanted to show the potential that machine learning has in regards to this area of interest. While we were not able to test a wide variety of health conditons or use more patients in this study, we have already showed promising findings that could be built further upon. By being able to detect early or new AMP presence within people, medical professionals will be able to take preventative action to ensure that someones condition does not possibly get worse over time.
In the future, we would like to add more health condtions and patients to help generalize our model and findings as time constraints limited us with how far we can expand. Additionally, we would like to incorporate more models such as the LSTM or BERT models to see if we can produce better and more accurate results.